
Синдром Бругада – генетически обусловленное кардиологическое состояние, характеризующееся различными нарушениями работы сердца, которые приводят к резкому повышению риска развития внезапной сердечной смерти. Симптомами этого состояния являются приступы пароксизмальной тахикардии, обмороки, фибрилляции предсердий и жизнеугрожающие фибрилляции желудочков, чаще всего возникающие во время сна. Диагностика синдрома Бругада производится на основании характерного симптомокомплекса, электрокардиографических данных и изучения наследственного анамнеза, некоторые формы патологии определяются молекулярно-генетическими методами. Специфического лечения заболевания не существует, применяют антиаритмическую терапию, используют разнообразные кардиостимуляторы.
Синдром Бругада – группа генетических нарушений, приводящих к изменению ионной проницаемости мембран кардиомиоцитов, вследствие чего возникают патологии ритма и проводимости, создающие повышенный риск внезапной сердечной смерти. Впервые такое состояние было описано в 1992 году двумя братьями – бельгийскими кардиологами испанского происхождения Хосе и Педро Бругада, обратившими внимание на взаимосвязь определенных электрокардиологических проявлений и нарушений сердечного ритма. В настоящее время установлено, что синдром Бругада является наследственным состоянием с предположительно аутосомно-доминантным механизмом передачи, удалось выявить несколько генов, мутации которых способны вызывать это заболевание. По некоторым данным, практически половина всех случаев внезапной сердечной смерти в мире обусловлены именно этой патологией. Распространенность синдрома Бругада различается в разных регионах планеты – в странах Америки и Европы она составляет примерно 1:10 000, тогда как в африканских и азиатских государствах это заболевание встречается чаще – 5-8 случаев на 10 000 населения. Синдром Бругада примерно в 8 раз чаще поражает мужчин, чем женщин, проявления патологии возникают в разном возрасте, но чаще всего выраженная симптоматика наблюдается в 30-45 лет.
Причины и классификация синдрома Бругада
Причина развития нарушений при синдроме Бругада заключается в патологической работе ионных каналов кардиомиоцитов, в основном натриевых и кальциевых. Их дефект, в свою очередь, обусловлен мутациями генов, кодирующих белки ионных каналов. Методами современной генетики удалось достоверно идентифицировать 6 основных генов, поражение которых приводит к развитию синдрома Бругада, в отношении еще нескольких существует подозрение, но отсутствует необходимая доказательная база. На этой основе построена классификация данного состояния, включающая в себя 6 форм заболевания (BrS):
• BrS-1 – наиболее распространенный и хорошо изученный вариант синдрома Бругада. Обусловлен мутацией гена SCN5A, расположенного на 3 хромосоме. Продуктом экспрессии данного гена является альфа-субъединица натриевого канала 5 типа, широко представленного в миокарде. Помимо синдрома Бругада мутации данного гена становятся причиной большого количества наследственных кардиологических патологий – семейной фибрилляции предсердий, синдрома слабости синусового узла 1 типа и ряда других.
• BrS-2 – данная разновидность синдрома Бругада вызывается дефектами гена GPD1L, который локализован на 3 хромосоме. Он кодирует один из компонентов глицерол-3-фосфат дегидрогеназы, принимающей активное участие в работе натриевых каналов кардиомиоцитов.
• BrS-3 – этот тип синдрома Бругада обусловлен дефектом гена CACNA1C, расположенного на 12 хромосоме. Продуктом его экспрессии является альфа-субъединица кальциевого канала L-типа, также присутствующего в кардиомиоцитах.
• BrS-4 – как и в предыдущем случае, причиной развития синдрома Бругада 4 типа является поражение потенциал-зависимых кальциевых каналов L-типа. Оно обусловлено мутацией гена CACNB2, расположенного на 10 хромосоме и кодирующего бета-2-субъединицу вышеуказанных ионных каналов.
• BrS-5 – распространенная разновидность синдрома Бругада, обусловленная мутацией гена SCN4B, локализованного на 11 хромосоме. Он кодирует белок одного из малых натриевых каналов кардиомиоцитов.
• BrS-6 – вызывается дефектом гена SCN1B, расположенного на 19 хромосоме. Во многом этот вариант синдрома Бругада схож с первым типом заболевания, поскольку в этом случае тоже поражаются натриевые каналы 5 типа. Ген SCN1B кодирует бета-1-субъединицу этого ионного канала.
Кроме того, в развитии синдрома Бругада подозреваются мутации генов KCNE3, SCN10A, HEY2 и некоторых других. Однако на сегодняшний день достоверно доказать их роль в возникновении данного заболевания не удается, поэтому пока количество генетических вариантов синдрома Бругада ограничено шестью. Наследование всех форм данной патологии неясно, лишь у 25% больных определяются признаки аутосомно-доминантной передачи. Предположительно имеет место доминантный тип наследования с неполной пенетрантностью либо влияние спонтанных мутаций. Также непонятны причины того, почему синдром Бругада чаше поражает мужчин, нежели женщин – возможно, выраженность проявлений заболевания находится в зависимости от гормонального фона больного.
Патогенез нарушений при любой форме синдрома Бругада примерно одинаков – из-за изменения проницаемости мембраны кардиомиоцитов для ионов натрия происходит нарушение трансмембранного потенциала и взаимосвязанных с ним характеристик возбудимых тканей: возбудимости, сократимости, передачи возбуждения окружающим клеткам. В результате возникают блокады проводящих путей сердца (пучков Гиса), тахиаритмии, усиливающиеся при повышении вагусных воздействий (во время сна). Степень выраженности симптомов при синдроме Бругада зависит от доли пораженных натриевых каналов. Усиливать проявления болезни могут некоторые лекарственные вещества, способны ингибировать ионные каналы сердца.
Симптомы синдрома Бругада
Возраст появления первых признаков синдрома Бругада сильно отличается у разных больных – были зарегистрированы случаи этой патологии как у детей 3-4 лет, так и у лиц старческого возраста. Одним из первых проявлений патологии становятся изменения на электрокардиограмме при полном отсутствии других клинических симптомов, поэтому данное заболевание нередко выявляется случайно. В большинстве случаев выраженная клиника синдрома Бругада возникает в возрасте 30-45 лет, этому предшествует бессимптомный период продолжительностью 10-12 лет, в течение которого единственным признаком патологии являются изменения на ЭКГ.
Обычно больные синдромом Бругада жалуются на беспричинное головокружение, обмороки, частые приступы тахикардии, особенно в ночное время или в период дневного отдыха. Иногда отмечается аномальная реакция на прием некоторых лекарственных средств – антигистаминных препаратов первого поколения, бета-адреноблокаторов, ваготонических средств. Их применение при синдроме Бругада может сопровождаться усилением побочных явлений, а также сердцебиениями, обмороками, падением артериального давления и другими негативными проявлениями. Никаких других симптомов при этом заболевании не выявляется, чем объясняется редкое обращение пациентов к кардиологу или другим специалистам – в ряде случаев проявления синдрома Бругада достаточно редкие и слабовыраженные. Тем не менее, это не снижает риск внезапной сердечной смерти, обусловленной данной патологией.
Диагностика синдрома Бругада
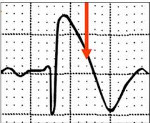
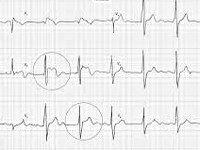
Для определения синдрома Бругада используют электрокардиографические методики, изучение наследственного анамнеза пациента, молекулярно-генетический анализ. Заподозрить наличие этого заболевания можно при наличии синкопальных явлений (головокружений, обмороков) неясного происхождения, жалоб на внезапные приступы тахиаритмий. Изменения на электрокардиограмме при синдроме Бругада могут определяться на фоне полного отсутствия клинических симптомов заболевания. При этом кардиологи выделяют три основных типа изменений на ЭКГ, незначительно отличающихся между собой. Типичная картина электрокардиограммы при синдроме Бругада сводится к элевации (подъему) сегмента ST над изоэлектрической линией и отрицательному зубцу Т на правых грудных отведениях (V1-V3). Также могут определяться признаки блокады правой ножки пучка Гиса, при холтеровском мониторировании выявляются приступы пароксизмальной тахикардии или фибрилляции предсердий.
Как правило, наследственный анамнез больных синдромом Бругада отягощен – среди родственников или предков имеются случаи летальных исходов от сердечной недостаточности, смерти во сне или внезапной сердечной смерти. Этот факт, а также наличие вышеперечисленных симптомов и изменений на ЭКГ дает основания для проведения молекулярно-генетической диагностики. В настоящее время врачи-генетики в подавляющем большинстве клиник и лабораторий производят определение синдрома Бругада, вызванного только мутациями генов SCN5A и SCN4B (1 и 5 типы патологии), в отношении остальных форм методы генетической диагностики пока не разработаны. Дифференцировать это состояние следует с реакцией организма на прием некоторых лекарственных средств, хроническим миокардитом и другими кардиологическими патологиями.
Лечение и профилактика синдрома Бругада
Специфических методов лечения синдрома Бругада на сегодняшний момент не существует, поэтому ограничиваются только борьбой с проявлениями этого заболевания, а также профилактикой жизнеугрожающих приступов тахиаритмии и фибрилляций. Наиболее широко при этом состоянии применяется амиодарон, несколько реже используются дизопирамид и хинидин. Однако медикаментозная терапия при синдроме Бругада в ряде случаев является малоэффективной, единственным надежным средством профилактики аритмии и внезапной сердечной смерти в этом случае становится имплантация кардиовертера-дефибриллятора. Только этот прибор способен оценивать работу миокарда больного и при патологических и жизнеугрожающих изменениях сердечного ритма приводить ее в норму посредством электрического разряда.
Многие традиционные антиаритмические препараты при синдроме Бругада противопоказаны, так как они угнетают деятельность натриевых каналов кардиомиоцитов и усиливают проявления патологии. К средствам, запрещенным при этом заболевании, относят аймалин, пропафенон, прокаинамид. Поэтому больным синдромом Бругада следует обязательно сообщать специалистам об имеющемся диагнозе, чтобы избежать назначения неверного антиаритмического средства. При наличии подобного заболевания у родственников или случаях внезапной сердечной смерти в роду следует регулярно производить ЭКГ-исследование для как можно более ранней диагностики этого состояния.
Прогноз синдрома Бругада
Прогноз синдрома Бругада неопределенный, так как степень выраженности симптомов заболевания очень вариабельна и находится в зависимости от ряда факторов. При наличии только электрокардиографических проявлений патологии без выраженных клинических симптомов прогноз относительно благоприятный. Если синдром Бругада сопровождается потерями сознания и приступами аритмии – без установки кардиовертера-дефибриллятора риск внезапной сердечной смерти возрастает во много раз. При применении данного прибора прогноз несколько улучшается, поскольку устройство может круглосуточно корректировать патологические изменения сердечного ритма.
Источник: http://www.krasotaimedicina.ru/diseases/genetic/Brugada-syndrome